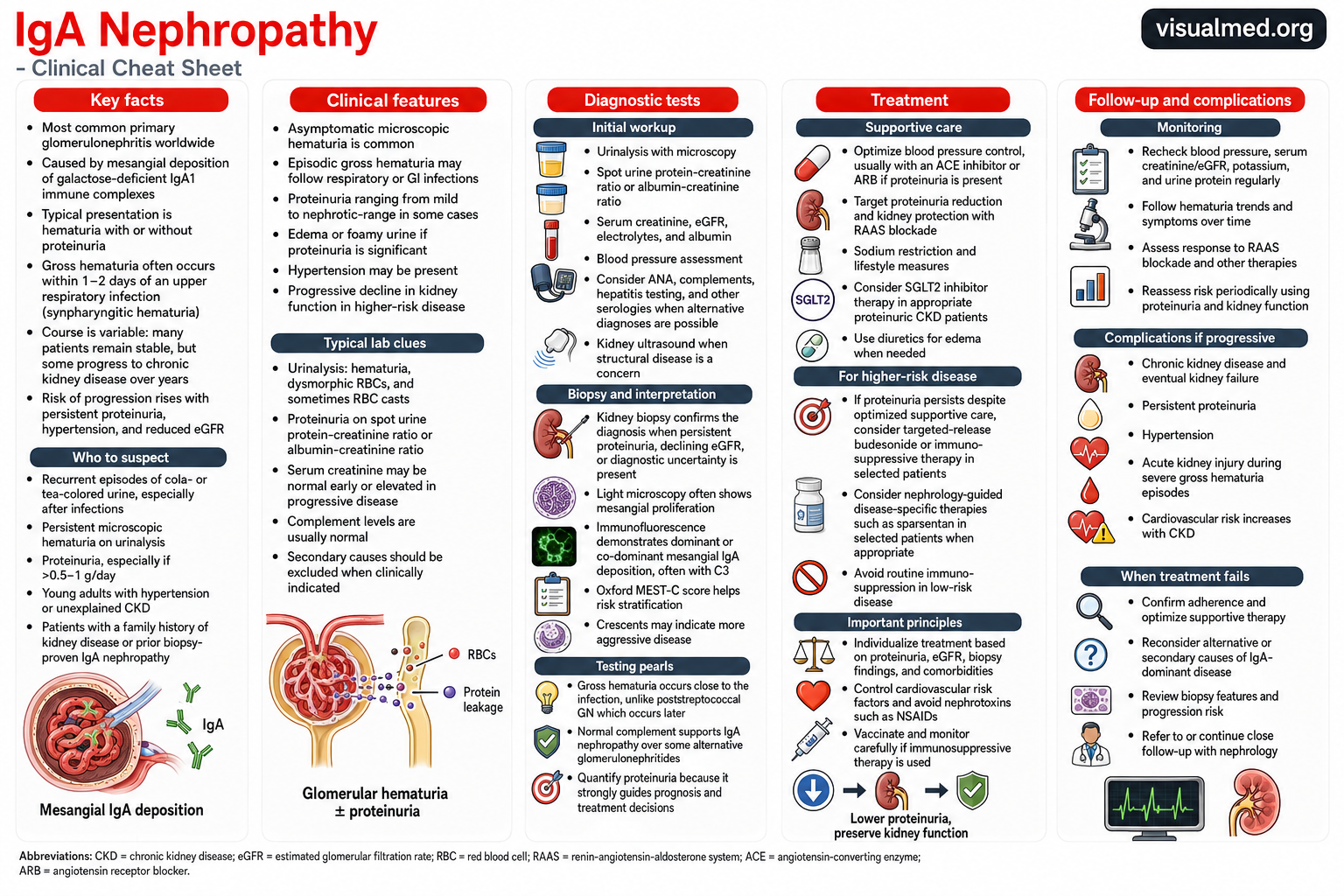
IgA nephropathy, also called Berger disease, is an immune-mediated glomerular disease caused by the accumulation of immunoglobulin A–containing immune complexes within the kidney’s filtering units. It is among the most common primary glomerular diseases worldwide and can range from an incidental urinary abnormality to progressive chronic kidney disease and kidney failure.
Although many patients remain clinically stable for years, persistent proteinuria, hypertension, and declining estimated glomerular filtration rate, or eGFR, identify individuals at greater risk of progression.
What Causes IgA Nephropathy?
IgA is an antibody that normally helps protect mucosal surfaces, including the respiratory and gastrointestinal tracts. In IgA nephropathy, abnormal forms of IgA1—commonly measured as galactose-deficient IgA1—participate in the formation of circulating immune complexes.
These complexes become deposited in the mesangium of the glomeruli. The resulting inflammatory response damages the glomerular filtration barrier, allowing red blood cells and protein to pass into the urine. Over time, repeated inflammation may lead to glomerular scarring, tubulointerstitial fibrosis, and progressive nephron loss.
Clinical Presentation
The presentation of IgA nephropathy is highly variable. Some patients have no symptoms and are diagnosed after microscopic hematuria or proteinuria is detected during routine testing.
A classic presentation is an episode of visible, cola- or tea-colored urine occurring during or shortly after an upper respiratory or gastrointestinal infection. This close temporal relationship is sometimes called synpharyngitic hematuria.
Other possible features include:
- Persistent microscopic hematuria
- Mild to nephrotic-range proteinuria
- Foamy urine
- Hypertension
- Swelling of the legs or around the eyes
- Progressive loss of kidney function
- Acute kidney injury during severe episodes of visible hematuria
Nephrotic syndrome is uncommon but may occur, sometimes when IgA deposition coexists with a podocytopathy resembling minimal change disease.
When Should IgA Nephropathy Be Suspected?
IgA nephropathy should be considered in patients with recurrent visible hematuria after infections, persistent microscopic hematuria, unexplained proteinuria, hypertension at a young age, or chronic kidney disease without another clear cause.
The risk of progression rises as proteinuria increases. KDIGO considers persistent protein excretion of at least 0.5 g per day an important marker of increased lifetime risk, even when kidney function is still relatively preserved.
Diagnostic Evaluation
Initial testing generally includes:
- Urinalysis with urine microscopy
- Urine protein-to-creatinine or albumin-to-creatinine ratio
- Serum creatinine and eGFR
- Serum electrolytes and albumin
- Blood-pressure measurement
- Kidney ultrasound when structural disease is suspected
Urine microscopy may demonstrate dysmorphic red blood cells or red blood cell casts, supporting a glomerular source of bleeding.
Additional blood tests may be used to exclude lupus nephritis, infection-related glomerulonephritis, hepatitis-associated disease, ANCA-associated vasculitis, and other causes of hematuria or proteinuria.
Kidney Biopsy
A kidney biopsy is required to confirm IgA nephropathy because no validated blood or urine biomarker can independently establish the diagnosis. The 2025 KDIGO guideline recommends considering biopsy in adults with proteinuria of at least 0.5 g per day when IgA nephropathy is a possible diagnosis and biopsy is not contraindicated.
Typical biopsy findings include:
- Mesangial proliferation on light microscopy
- Dominant or co-dominant mesangial IgA deposits on immunofluorescence
- Frequent accompanying C3 deposition
- Variable segmental sclerosis, tubular atrophy, interstitial fibrosis, or crescents
The Oxford MEST-C classification describes mesangial hypercellularity, endocapillary hypercellularity, segmental sclerosis, tubular atrophy or interstitial fibrosis, and crescents. These findings assist with risk assessment but should be interpreted alongside proteinuria, blood pressure, eGFR, and the clinical course.
Treatment Goals
Treatment aims to reduce proteinuria, control blood pressure, prevent additional glomerular injury, and slow the decline in kidney function.
For patients at risk of progression, KDIGO recommends maintaining urine protein excretion below 0.5 g per day, and ideally below 0.3 g per day, when achievable. The long-term goal is to reduce eGFR decline toward the physiological rate expected with aging.
Supportive Kidney-Protective Therapy
Supportive care remains the foundation of treatment.
ACE Inhibitors or ARBs
An angiotensin-converting enzyme inhibitor or angiotensin receptor blocker is generally used at the maximally tolerated dose, unless contraindicated. These medications reduce intraglomerular pressure, lower proteinuria, and provide long-term kidney protection.
ACE inhibitors and ARBs should not ordinarily be taken together. Kidney function and potassium should be checked after starting therapy or increasing the dose.
Blood-Pressure and Lifestyle Management
KDIGO emphasizes strict blood-pressure control together with sodium restriction, weight management, regular exercise, smoking cessation, and management of cardiovascular risk factors. The guideline proposes a blood-pressure target of approximately 120/70 mm Hg or lower when safely tolerated, although targets must be individualized.
Patients should also avoid unnecessary nephrotoxins, particularly regular NSAID use.
SGLT2 Inhibitors
SGLT2 inhibitors may be considered in patients with IgA nephropathy who are at risk of progressive kidney-function loss, particularly those with proteinuric chronic kidney disease. These agents may reduce intraglomerular pressure and slow kidney decline, including in people without diabetes.
The evidence is less certain in younger patients with preserved eGFR and few additional risk factors, so treatment should be individualized.
Disease-Specific Treatment
Patients with ongoing proteinuria or declining kidney function despite optimized supportive care may require disease-specific therapy under nephrology supervision.
Targeted-Release Budesonide
A targeted-release formulation of budesonide, known as Nefecon or Tarpeyo in some markets, acts primarily within the distal small intestine, where mucosal immune activity is thought to contribute to abnormal IgA production.
KDIGO suggests a nine-month course for selected patients at risk of progressive disease. The durability of the response and the role of repeated courses continue to be evaluated.
Sparsentan
Sparsentan is a dual endothelin and angiotensin receptor antagonist that reduces proteinuria and slows kidney-function decline in selected adults with primary IgA nephropathy at risk of progression. Because it already provides angiotensin-receptor blockade, it should not be combined with an ACE inhibitor or ARB.
Systemic Glucocorticoids
Reduced-dose systemic glucocorticoids may be considered in selected higher-risk patients when targeted-release budesonide is unavailable or unsuitable. Treatment requires careful assessment because steroids can cause serious adverse effects, including infection, hyperglycemia, weight gain, osteoporosis, and gastrointestinal complications.
The risk of toxicity is particularly important in patients with diabetes, obesity, advanced chronic kidney disease, latent infection, osteoporosis, or active peptic-ulcer disease.
Routine immunosuppression is not appropriate for patients with low-risk or stable disease.
Monitoring and Follow-Up
Long-term follow-up is essential because IgA nephropathy can progress silently.
Monitoring usually includes:
- Blood pressure
- Serum creatinine and eGFR
- Serum potassium
- Urine protein quantification
- Hematuria trends
- Medication tolerance and adherence
- Cardiovascular risk factors
The frequency of monitoring depends on the amount of proteinuria, baseline kidney function, rate of eGFR decline, treatment regimen, and biopsy findings.
Potential Complications
Progressive IgA nephropathy may lead to persistent proteinuria, hypertension, chronic kidney disease, acute kidney injury during severe hematuria, and eventually kidney failure requiring dialysis or transplantation.
Patients with chronic kidney disease also face increased cardiovascular risk. Early recognition, proteinuria reduction, blood-pressure control, and regular nephrology follow-up are therefore central to improving long-term outcomes.
Key Takeaway
IgA nephropathy should be suspected in patients with recurrent infection-associated hematuria, persistent microscopic hematuria, or otherwise unexplained proteinuria. Kidney biopsy establishes the diagnosis, while proteinuria and eGFR are among the most important markers used to guide treatment and follow-up.
Modern management combines intensive supportive kidney care with disease-specific therapy for appropriately selected higher-risk patients. Treatment should always be individualized in consultation with a nephrologist.
This article is intended for medical education and does not replace individualized clinical assessment or treatment advice.